
 Extracting and comparing gap regions
Extracting and comparing gap regions
10a. MASK
The MASK program extracts a specified set of gap regions from a
.srf file and writes them out to a file called mask.srf. It
can also write out a file in one of the standard molecular graphics formats
(eg QUANTA, InsightII, etc.). It is
particularly useful in selecting a protein's binding site from all its
internal cavities and surface clefts.
Optionally, the program can also write out all atoms within a preset
distance of the selected gap regions. This may be useful for pulling out
just those atoms making up the surface of a binding site.
MASK can only be run on a .srf file of type G. That
is, a gaps-file generated to contain either the gap-regions between two
molecules (see Gaps between molecules) or the
clefts and cavities in a single molecule (see Clefts
and cavities). In both cases, SURFNET will have written out two
additional files: gaps.pdb and gaps.pnt.
The gaps.pdb file
The gaps.pdb file is a PDB-format file in which each gap
region generated by SURFNET is represented by a single ATOM
record. Each atom is located at the centre of mass position of the
corresponding gap region, and the atoms are listed in descending order of
gap-volume (which is shown on the right of the record). Being a
PDB-format file, it can be viewed on any molecular graphics
package. When superimposed on the gap regions generated by SURFNET
the atoms allow you identify which gap region is which (eg by
clicking on the relevant atom you can get the atom's residue number and
hence the number of the corresponding gap region).
A protein's binding site - which in most cases has the largest volume - will
usually be represented by the topmost ATOM record.
(The gaps.pnt file is a binary file used by SURFNET to hold
the pointers relating the grid-points in the gaps.srf file to each
of the different gap regions).
Running MASK
Run MASK as follows:-
- Identify which gap-region(s) you want extracted, say by viewing the
gaps.pdb file on the graphics, along with the gap regions
themselves, as described above.
- Edit the gaps.pdb file to remove all ATOM records except
those corresponding to the gap regions you want extracted.
- Run MASK by typing: mask. You will be asked:-
- Enter name of input .srf density file. This will usually be
gaps.srf.
- Enter map-format required: (Q)uanta, (C)CP4, (S)ybyl, (I)nsightII or
(N)one. Enter whichever is appropriate.
- Are neighbouring atoms required for mask region - (Y/N)?. If you
answer Y, the program will extract all atoms within a preset
distance of the selected gap regions and write them out to a file called
cavatoms.pdb. You will first be asked:-
- Enter name of corresponding PDB file. Enter the name of the
PDB file from which the original gap regions were generated.
- Enter cut-off distance between atoms and the mask region (eg
4.0). Enter how close the atoms need to be to the gap regions in order
to be written out to cavatoms.pdb (usually 4.0Å is adequate).
The output file containing the extracted gap regions is called
mask.srf. If the neighbouring atoms option has been selected, these
atoms will be written to cavatoms.pdb. You can then run
SURFNET on the cavatoms.pdb file to give the surface of these
atoms. When viewed on the graphics, either in a different colour from the
rest of the protein's surface, or as a solid, shaded surface, this gives a
good depiction of the binding site's actual surface.
The two examples below show the largest gap region for endothiapepsin,
PDB code 3er5 (see Clefts and
cavities). This largest cleft corresponds to the protein's binding site
and both plots show how the inhibitor molecule sits within the cleft,
hanging out at both ends, particularly on the right-hand side.
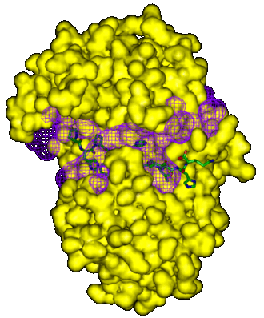
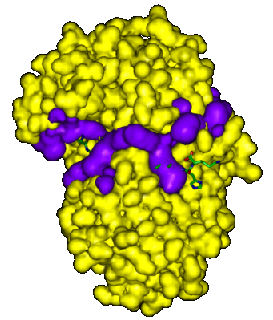
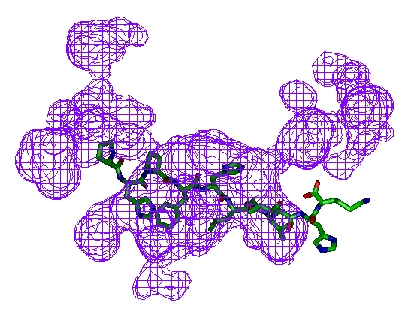
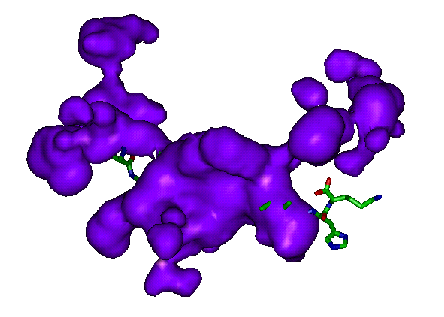
The examples below show close-ups of just the binding site, with the
protein removed.
All examples given here were rendered using InsightII.
Extracting and comparing gap regions